Technology review
Antibody-drug conjugates (ADCs) are a biotherapeutic modality typically used for the treatment of cancer. ADCs comprise a monoclonal antibody (mAb) conjugated via a linker to a cytotoxic drug or payload to facilitate targeted or ‘smart’ chemotherapy. Once bound to a cancer cell receptor, the ADC is internalised and releases the payload into the cancer cell causing cell death. This targeted delivery minimises off-target toxicity, potentially reducing side effect severity experienced with traditional chemotherapy. Properties of the antibody (target specificity), linker (mode and time of linker cleavage, such as by proteases or pH changes), and stability or potency of the cytotoxic drug all need to be carefully considered in the development of an effective ADC to treat a selected cancer.
Indications treated
ADCs were the first biotherapeutic modality to be approved in the 21st century, with the FDA giving the green light for gemtuzumab ozogamicin (Mylotarg®) in 2000 for use in patients with relapsed acute myelogenous leukaemia (AML). This drug targets the CD33 receptor expressed on leukemic myeloblasts, and upon internalisation releases the DNA-cleaving drug calicheamicin. Almost a quarter-century later, new ADCs are still being developed and approved for use. The most recent ADC to receive FDA approval as of January 2025, was Datopotamab Deruxtecan (Dato-DXd) for the treatment of certain patients with unresectable or metastatic hormone receptor-positive, HER2-negative breast cancer. Before that, Tivdak® (Tisotumab vedotin) was approved in August 2024 for the treatment of patients with recurrent or metastatic cervical cancer, utilising the microtubule-disrupting monomethyl auristatin E (MMAE) drug and an anti-tissue factor antibody. One current ADC research avenue involves reducing the size of ADCs to allow for greater penetration efficiency by using minibodies or single-chain variable fragment regions, such as PEN-221 in treating small cell lung cancer (Whalen et al., 2019).
Patent considerations
Considerable investment is required to take an ADC from conception to market, so delaying biosimilar competition is important for recouping that investment. While there are technical challenges in producing biosimilar ADCs, and some regulatory exclusivities available, effective patent protection is essential, be it for driving investment in the early stages of a product’s life, to deterring biosimilars in the later stages. Given the right strategy, many innovative aspects of ADCs may be patentable, including the specific antibody or payload, appropriate compositions and formulations of the conjugate, methods of manufacture, and medical uses. Since the targeting property of ADCs provides them with their main therapeutic advantage, the development of new antibodies and antibody fragments is the main driver in the development of new therapeutic ADCs. This means special considerations for antibody patents must be taken into account, which we have discussed in an earlier article in this series.
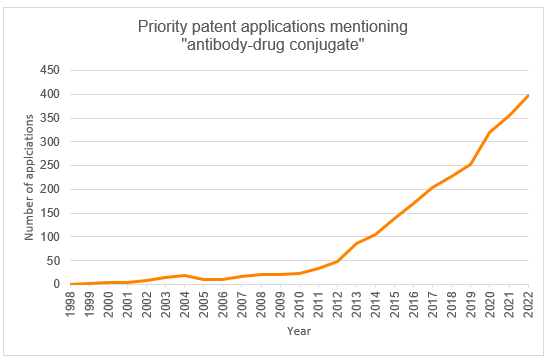
Priority patent applications filed worldwide mentioning “antibody-drug conjugate” have seen a steady increase since the inception of the technology (Figure 1). We predict that this trend will continue owing to further developments in precision targeting, a rise in cancer incidence, and a continued momentum from new regulatory approvals (Patbase, 2024). Cumulatively, the top filers are Genentech, Seagen, and Regeneron Pharmaceuticals.
When patenting antibodies in Europe, there are several options for how an antibody may be claimed. Antibodies may be defined, by their structural features (amino acid or coding nucleotide sequences), functional features (by virtue of their target antigen or epitope or other characteristics), or their production process (including the hybridoma producing the antibody). Not all of these are possible outside of Europe where the requirements diverge considerably. It is therefore important to consider the different approaches across the globe when drafting new patent applications for antibodies, as with many other types of biotech inventions.
As with any invention, an antibody claim must meet, amongst other things, the requirements of novelty and inventive step. How an antibody is claimed will depend on both the structure and function of known antibodies, and what contribution and advantage the new antibody provides, as evidenced by experimental data. Recent case law in Europe has emphasised the need for supporting data in patent applications for these technologies. The advantage of the new antibody relied upon may be a result of complementarity-determining regions (CDR) sequences of the antibody, or possibly a result of the specific epitope on the target, the latter of which may justify a broader claim towards the target rather than a narrower claim to the antibody amino acid sequence. Also, some novel antibody modifications may provide advantages that apply to multiple different antibodies, such as constant region modifications that reduce adverse immune reactions to the antibody or enhance the antibody’s efficacy. Such platform inventions require a different drafting strategy. Every instance is different but generally speaking, broader claims will require more data and more examples of appropriate antibodies to be provided in the patent application to meet the requirements of patentability. This is particularly the case for sufficiency of disclosure, which necessitates that the patent application must describe the invention in enough detail so that it can be reproduced across the claimed scope.
In cases where a new antibody binds a known target that existing antibodies bind to, a broad claim to an antibody defined by the target (e.g. “an antibody binding to [protein X]”) would not be novel but a claim to an antibody defined by a more narrowly defined novel epitope (e.g., “an antibody binding to [amino acids 10 to 20 of protein X]”) may be novel. If this is not novel, then it may be necessary to define the antibody by its own structure, rather than its target. For example, it could be defined by its CDRs with or without the framework region, or indeed by the full variable regions, or even the sequence of the whole antibody, depending on the circumstances. However, a novel claim at the EPO is unlikely to be considered inventive unless the application includes some data showing a special advantage, either of the antibody or the target, that is not provided by known antibodies. This may be, for example, increased therapeutic activity, enhanced stability, or decreased immunogenicity, the effect of which can be attributed to CDR sequences or a specific epitope. Claim scope and breadth of data/examples come hand in hand: typically the more data/examples, the broader the justified claim scope. Usually, data can be provided after filing to support inventive step so it doesn’t always need to be available when drafting the patent application.
EPC Board of Appeal decision T 0835/21 concerned the sufficiency and inventive step of an antibody defined as binding to a particular epitope on human LRP6 and inhibiting Wnt3/Wnt3a signalling activity. The application identified just two Fabs that had the indicated property, but they were not reproducible as no sequence data was provided. The Board determined that, although the specific antibodies were not reproducible, there was enough information for new antibodies with the indicated properties to be produced so the claim was sufficient, and the Wnt antagonism was unexpected so inventive. While claims were allowed in this case with no sequence limitations, this is often not the case in Europe. As an interesting aside, the situation is very different in the US. Following Amgen Inc. v. Sanofi, 598 U.S. 594 (2023), it is now a requirement of US antibody claims to specify the antibody sequence, regardless of the number of exemplified antibodies. US antibody patents granted after this decision are now all limited by at least their CDR sequences, and while broader patents granted before this decision may be in force, they may not withstand an invalidity challenge in the current climate.
Regarding antibody-drug conjugate case law before the EPO, it appears just two Board of Appeal decisions mention this biotherapeutic modality. T 2517/19 held that an inventive step was present due to the identification of a specific residue where a cysteine may be introduced to conjugate duocarmycin, rather than to a native cystine, to reduce hydrophobicity and increase clearance times. The prior art disclosed a general region where cystine engineering may be appropriate based on distance to the antigen binding site and thiol reactivity that encompassed this specific residue position but was absent of any teaching or indication towards reducing hydrophobicity. In T 0885/21, opponents had previously argued that the claims towards an ADC for use as a medicament would cover conjugates unsuitable for medical use due to broad definitions encompassing ADCs which lacked efficacy or were toxic, thereby insufficiently disclosing the invention and imparting undue burden on the skilled person in selecting working embodiments. However, the Board disagreed that a lack of sufficiency was present as the purpose-limited claims would functionally exclude any conjugates unsuitable for medical use, and that the patent provided sufficient guidance to carry out the claimed invention.
Summary
ADCs are a multi-faceted technology that spans both biologics and pharmaceutical chemistry. Bringing this class of drug to market requires careful patenting considerations to help achieve the strongest protection for a product. While the allowability of certain antibody claims is not consistent between jurisdictions, is it crucial that any application contains as much data as possible, such as defining the antibody in multiple ways and providing many experimental examples, to allow the broadest protection in each jurisdiction sought. When looking to patent antibodies and ADCs in Europe and the US, an up-to-date and detailed knowledge of the requirements is crucial.
The biotechnology group at GJE has extensive experience in patenting a diverse range of biotechnological inventions in addition to advising innovative biotech companies and investors. To discuss your biotech IP strategy, please contact us at biotech@gje.com.
References
Norsworthy et al. (2018). FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist. 23(9): 1103-1108. Available online, accessed July 2025.
Datopotamab Deruxtecan. Available online, accessed July 2025.
Whalen et al. 2019. Targeting the Somatostatin Receptor 2 with the Miniaturized Drug Conjugate, PEN-221: A Potent and Novel Therapeutic for the Treatment of Small Cell Lung Cancer. Mol Cancer Ther. 18(11): 1926-1936. Available online.
Genmab. TIVDAK® (tisotumab vedotin-tftv) Receives U.S. FDA Approval to Treat Recurrent or Metastatic Cervical Cancer. Available online, accessed July 2025
T 0835/21 – Available online.
T 2517/19 – Available online.
T 0885/21 – Available online.
Related resources

GJE announces alliance with Wächtershäuser & Hartz
GJE is pleased to announce that the firm has formed an alliance with the German patent and trade...

Key considerations when patenting gene editors in Europe
We set out the key things for innovators and applicants to consider when patenting gene editors in Europe.
Key considerations when patenting gene therapies in Europe
Technology review The impact of gene therapies has been revolutionary to the treatment of genetic disorders and diseases....